

В лабораториях обработка результатов секвенирования состоит из множества запусков специальных программ. Эти шаги организуют в виде конвейера действий (pipeline), в который на входе подаются FASTQ-файлы, а на выходе получают VCF-файл. Или таблицу, удобную для анализа человеком.
При помощи Galaxy и snpEff мы пройдем все эти шаги поэтапно, каждый раз останавливаясь и проверяя, что же мы получили.
Скорее всего, у вас уже есть готовый VCF-файл. Поэтому, если вы хотите сразу перейти к анализу генетических нарушений, то можно пропустить следующую главу о выравнивании и перейти к главе об аннотировании.
Но откуда вообще может возникнуть необходимость пройти весь путь от FASTQ до VCF-файла?
1) VCF-файла может не быть, если лаборатория не проводит постобработку данных.
2) Если вы проводили секвенирование несколько лет назад, то данные в BAM и VCF-файлах могли быть выровнены по референсному геному GRCh37/hg19, который не совместим со многими новыми базами данных.
3) В VCF-файл включаются не все варианты, некоторые отфильтровываются по тем или иным критериям. Кроме того, у лаборатории могут быть свои стандарты обработки и вывода данных, которые создадут проблемы при последующем анализе.
Важно учитывать, что эталонный геном (референсный геном) , с которым происходит сравнение ваших данных, нечасто, но уточняется. В версии эталонного генома под номером GRCh38/hg38, которая вышла в 2013 году и постепенно внедрилась, порядковый номер аллеля на хромосоме уже не советует предыдущей версии GRCh37/hg19.
Это очень важный момент для анализа!
Различные версии референсных геномов означают, что в дальнейшем нам нужно будет выбирать соответствующие версии баз данных. Иначе большая часть строк будет пропущена при аннотировании – алгоритмы просто не смогут увязать вместе позиции в геноме и базе данных.
Поэтому, если у вас есть готовый VCF-файл, и вы не хотите проходить весь путь, описанный в предыдущей главе, то нужно выяснить, какая версия генома использовалась при его создании. Для этого достаточно заглянуть в шапку VCF-файла и найти строку с номером сборки. Например, если вы увидите «assembly=hg19», то у вас старый референсный геном GRCh37/hg19. Если же вы увидите hg38, то у вас новый референсный геном GRCh38/hg38.
Содержание статьи
Выравнивание данных в Galaxy: от FASTQ к VCF-файлу
Прочитанные секвенатором Illumina последовательности ДНК обычно хранятся в нескольких (двух и более) парных FASTQ файлах, упакованных в архив (расширение fastq.gz).
Поскольку работа с FASTQ-файлами очень ресурсоемкая, к тому же требует много времени, мы воспользуемся мощной и бесплатной веб-платформой Galaxy. Вы можете прочитать эту официальную и подробную инструкцию, но в ней указаны не все шаги, а о некоторых правильных действиях приходится догадываться.
Поэтому я описал свои действия здесь и снял их на этом видео.
Рекомендую параллельно использовать текстовое описние ниже, видео и официальные инструкции.
Зарегистрируемся и подтвердим в email аккаунт на сайте Galaxy https://usegalaxy.org/
Автоматически созданный проект Unnamed history можно переименовать, например, в NGS results, через меню User -> Histories.
А затем выбрав в выпадающем списке Rename.

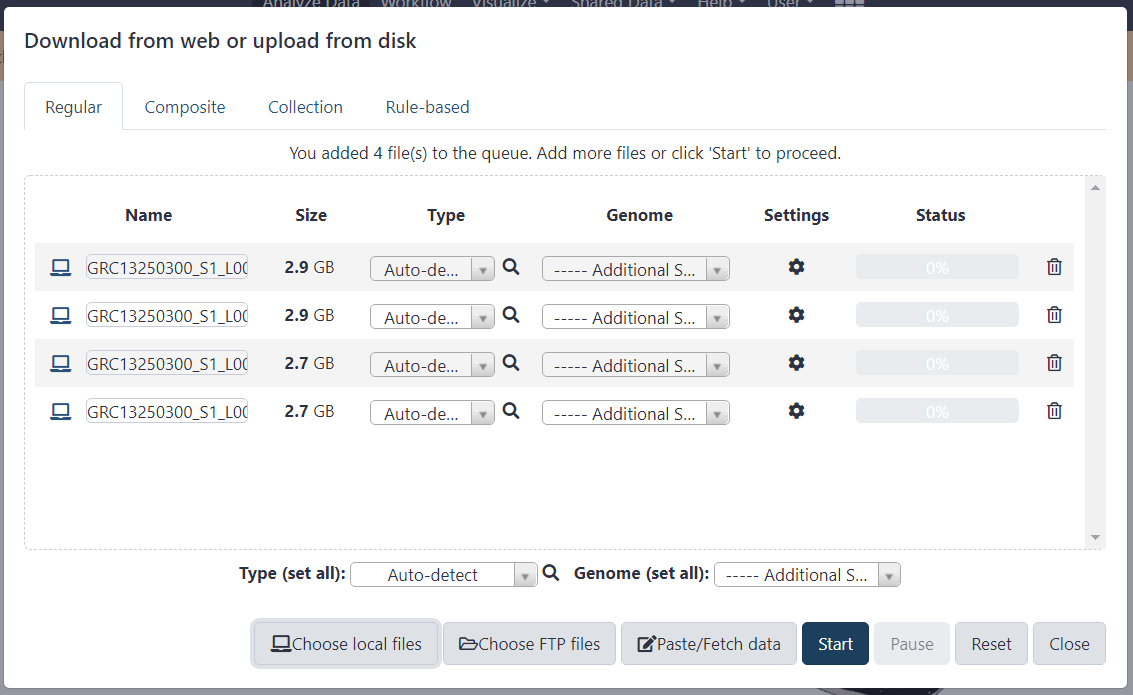
Загрузим наши упакованные файлы на сервер, кликнув иконку загрузки:

Дождемся пока файлы загрузятся и появятся в правой колонке подсвеченные зеленым фоном.
Прежде чем приступать к анализу, наверняка будет интересно проверить качество секвенирования, оценив данные в FASTQ файлах. Для проверки качества введите в строке поиска «fastqc». И выберите из выпадающего списка инструмент FastQC Read Quality reports.
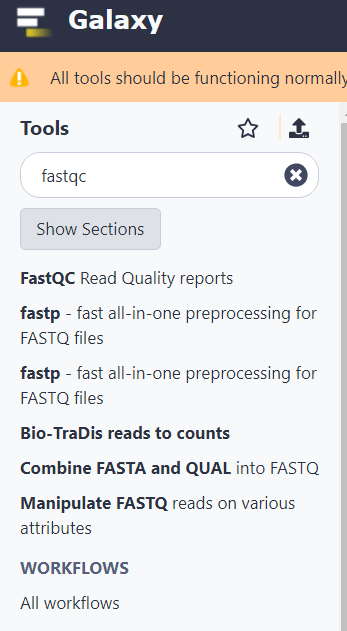
В верхнем поле «Short read data from your current history» нужно указать на иконку выбора нескольких файлов и потом на предварительно загруженные FastQ файлы. Затем нажать «Execute».
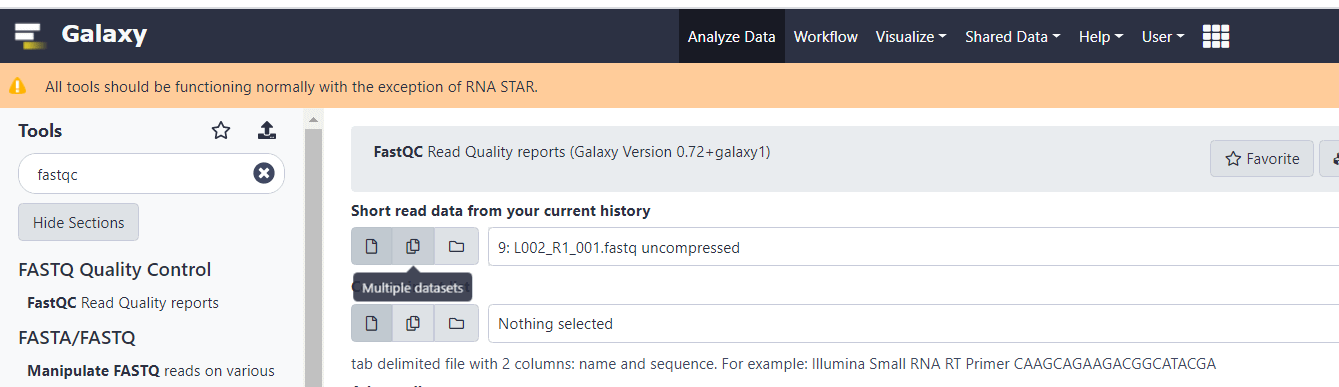
Прежде чем приступать к анализу, наверняка будет интересно проверить качество секвенирования, оценив данные в FASTQ файлах.
Секвенирование Illumina основано на идентификации отдельных молекул ДНК. У каждой из четырех молекул будет своя длина волны. Но из-за несовершенства процесса секвенирования и ограничений оптических инструментов, считывание (т.е. базовый вызов или base calling) всегда будет иметь не точный, а вероятностный характер. По этой причине файлы FASTQ хранят вместе с каждым считанным нуклеотидом, показатель качества – вероятность ошибки.
В биоинформатике используется статистическая функция phred quality score, которая определяет, насколько вероятно, что отдельный прочитанный нуклеотид (базовый вызов) может быть неверным. Например, оценка Phred 10 соответствует одной ошибке в каждых 10 базовых вызовах или точности 90%; оценка Phred 20 соответствует одной ошибке в каждых 100 базовых вызовах, или точности 99%.
Я посмотрел разницу между примерами качественных, некачественных и моих данных. Судя по зеленому флажку, мои данные приемлемого качества – лишь небольшая часть базовых вызовов приближается к Phred 20.



Но дальнейшие действия должны быть не произвольными, а одновременными для пар файлов.
Парными FASTQ-файлы называют потому, что чтение отрезков ДНК происходит с двух концов. Прямое и обратное прочтение сохраняются каждое в отдельном файле.

Направление прочтения должно быть указано в имени файлов. У файлов с прямым прочтением будет имя похожее на ***forvard.fastq.gz, а у файлов с обратным прочтением будет имя ***reverse.fastq.gz. Или различные направления будут обозначаться буквами R1 и R2.
Например, четыре наших файла с результатами секвенирования выглядели как в первом примере:
R1, f, forward – прямое прочтение
R2, r, reverse – обратное прочтение
Пример 1.
GRC13250*00_S1_L001_R1_001.fastq.gz – прямое прочтение, первая часть.
GRC13250*00_S1_L002_R1_001.fastq.gz – прямое прочтение, вторая часть
GRC13250*00_S1_L001_R2_001.fastq.gz – обратное прочтение, первая часть.
GRC13250*00_S1_L002_R2_001.fastq.gz – обратное прочтение, вторая часть.
Пример 2.
sample1-f.fq.gz – прямое прочтение, первая часть.
sample1-r.fq.gz – обратное прочтение, первая часть.
sample2-f.fq.gz – прямое прочтение, вторая часть
sample2-r.fq.gz – обратное прочтение, вторая частьВажно разобраться и не путать, в каких файлах находится прямое, а в каких обратное направление прочтения.
Если есть сомнения, не допустили ли вы ошибку с выбором пары, то лучше сразу отменить задание и создать его повторно, поскольку времени на его выполнение может уйти много, а ошибку мы выявим уже при работе с VCF-файлом.
Если мое объяснения не все прояснило, посмотрите видео главы «Mapping against a pre-computed genome index» по ссылке.
Далее очистим данные при помощи инструмента Trimmomatic, обрезая так называемые «адаптеры». Это служебный ДНК, который был необходим для проведения секвенирования Illumina.
Вводим в строке Trimmomatic, выбираем в выпадающем меню «Single-end or paired-end reads?» строку «Paired-end (two separate input files)»
Выбираем первый файл прямого и первый файл обратного прочтения среди загруженных файлов. Нажимаем «Execute».
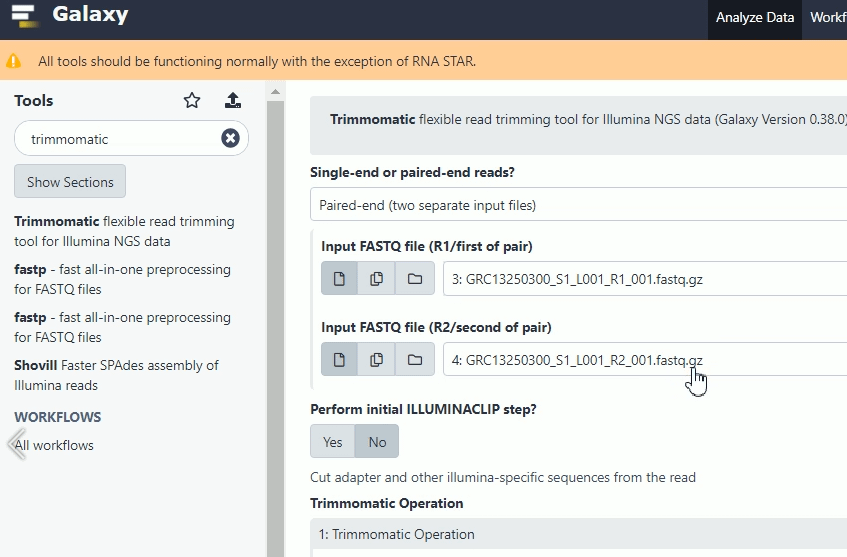
Аналогично поступаем для следующих пар.
Мы получили пары очищенных файла с префиксом Trimmomatic.
Теперь можно выровнять парные файлы по эталонному геному и получить BAM файлы, в которых все прочитанные нуклеотиды будут выстроены по порядку.
Наберем в строке поиска «BWA-MEM», затем в меню поиска выберем строку «Map with BWA-MEM – map medium and long reads (> 100 bp)». Затем в строке «Using reference genome» наберем 38 и выберем «Human (Homo sapiens) (b38): hg38». То есть выберем последнюю версию референсного генома.
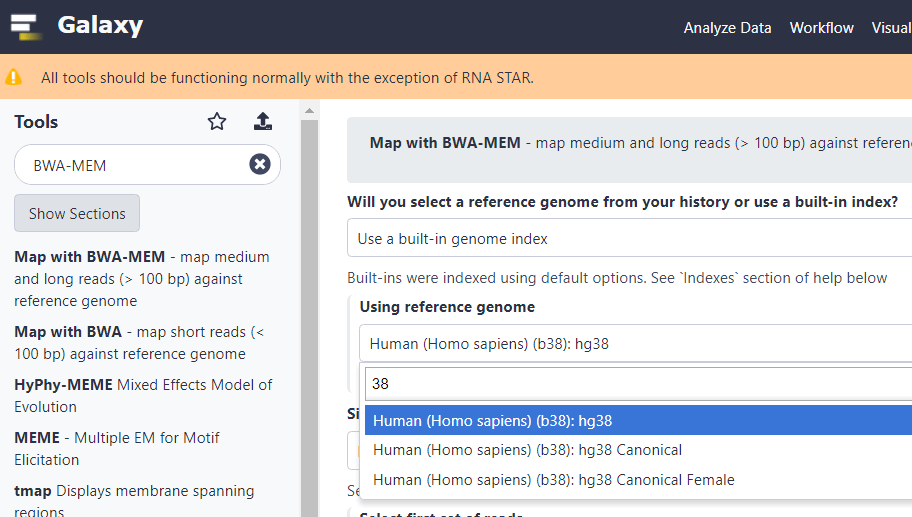
Важно! Если вы знаете, что вам придется использовать старые базы данных (где указаны кода GRCh37 и hg19), тогда для совместимости вам нужна предыдущая версия. В этом случае, здесь и далее наберите hg19 и выберите «Human (Homo sapiens) (b37): hg19». Иначе лучше использовать сборку hg38.
В поле «Single or Paired-end reads» указываем «Paired».
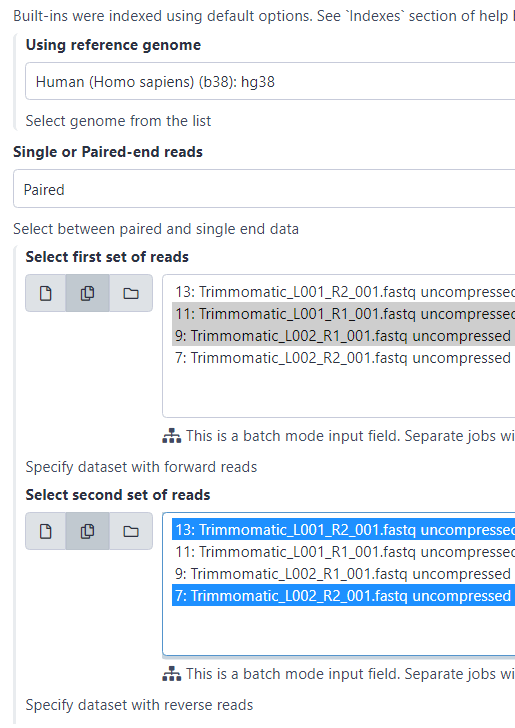
После чего появятся два поля с текстом «Select first set of reads» и «Select second set of reads». Возле них нужно выбрать иконки сдвоенных файлов, если у вас более чем два FASTQ-файла. При этом мы выбираем не исходные, а очищенные файлы с префиксом Trimmomatic.
В первом поле (кликая по порядку возрастания номеров!) выбираем файлы прямого чтения (с обозначением forward или R1), зажимая при выборе файлов клавишу ctrl.
Затем во втором поле, тоже по порядку возрастания, выбираем файлы обратного чтения (обозначения reverse или R2). Нажимаем «Execute».
Мы получим один или несколько выровненных BAM файлов, по числу файлов архива. Их имена будут начинаться со слов «Map with BWA-MEM on data…».
Объединим BAM-файлы в один.
Наберем в строке поиска «MergeSamFiles», затем в меню поиска выберем строку «MergeSamFiles merges multiple SAM/BAM datasets into one».
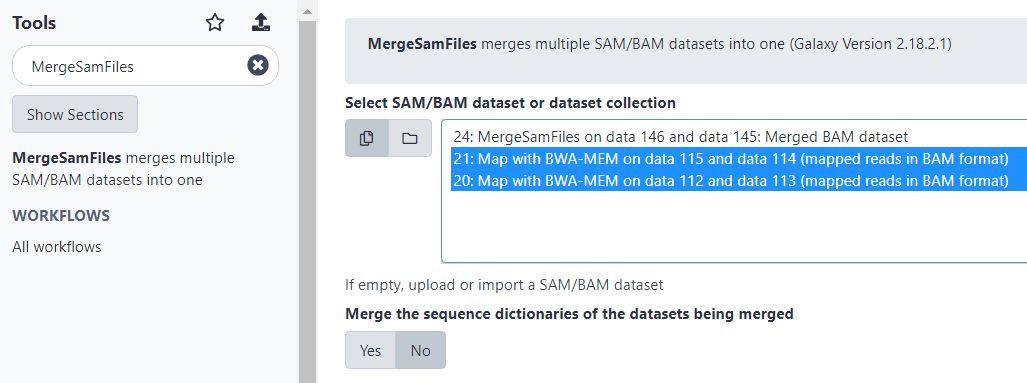
Затем в поле «Select SAM/BAM dataset or dataset collection» выберем два выровненных BAM-файла «Map with BWA-MEM on data…» и выполним команду.
В итоге должен получиться третий объединенный BAM-файл, который начинается с префикса «MergeSamFiles…».
Мы выровняли наш геном по эталону. А теперь мы выявим отличия нашего генома от эталона. Процесс сравнения называется variant calling. Те прочтения, которые отличаются от эталонного генома, записывают в VCF-файл. (Аббревиатура VCF означает variant call format).
Variant call мы будем делать при помощи мощного статистического детектора «FreeBayes».
Найдем его через строку поиска.

Здесь нам нужно указать на объединенный BAM-файл «MergeSamFiles…» и выбрать референсный геном «Human (Homo sapiens): hg38» (или же hg19, если вы пользуетесь не новыми, а старыми базами данных).
На выходе мы получим довольно большой VCF-файл, который начинается с префикса FreeBayes. Для экзома он содержит несколько миллионов строк, множество из которых будут с низким числом прочтений. Как правило, это прочтения за пределами кодирующих участков гена, которые не имеют особой ценности.
Отфильтруем прочтения низкого качества при помощи инструмента SnpSift Filter. На вход к нему подаем файл FreeBayes. Здесь в поле «Filter criteria» вставим текст «( QUAL > 30 ) & ( DP > 20 )».
То есть мы отсекаем прочтения с качеством по шкале Phred менее 30. Наверное, стоит сказать, что если мы используем FreeBayes, то в фильтре QUAL нет большого смысла. Потому что каждое прочтение будет вносить свой вклад в качество, а в итоге оно будет высоким и неинформативным. Но, возможно, мы отсечем какие-то совсем некачественные варианты.
Также отсечем прочтения с покрытием менее 20 (для экзома такая глубина отсечения кажется подходящей).
Количество записей относительно исходного VCF-файла (FreeBayes) сокращается более чем в 10 раз.
VCF-файл содержит, в общем, понятные аннотации относительно гомо- или гетерозиготности варианта (0/1, 1/1). Также, сравнивая фрагмент эталонного и альтернативного варианта, можно понять какой он – SNP, индель или сложная замена. Однако, для облегчения последующего анализа, можно добавить и более понятные аннотации с помощью функции SnpSift Variant Type.
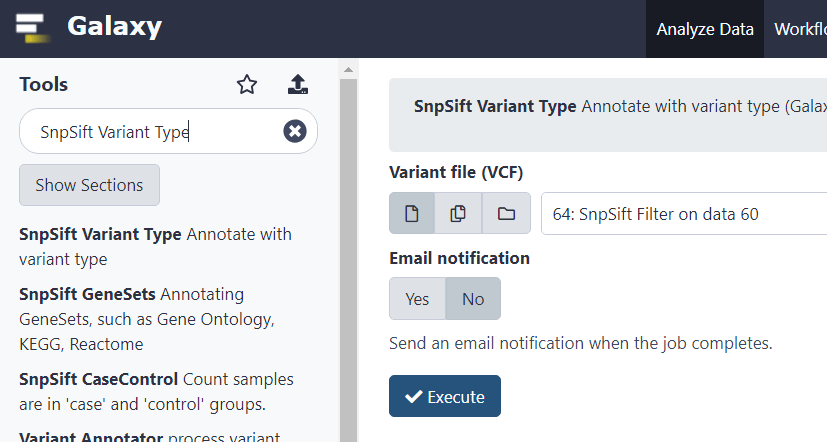
Далее аннотируем VCF-файл записями из базы dbSNP, чтобы упростить себе задачу и не делать этого на локальном компьютере.
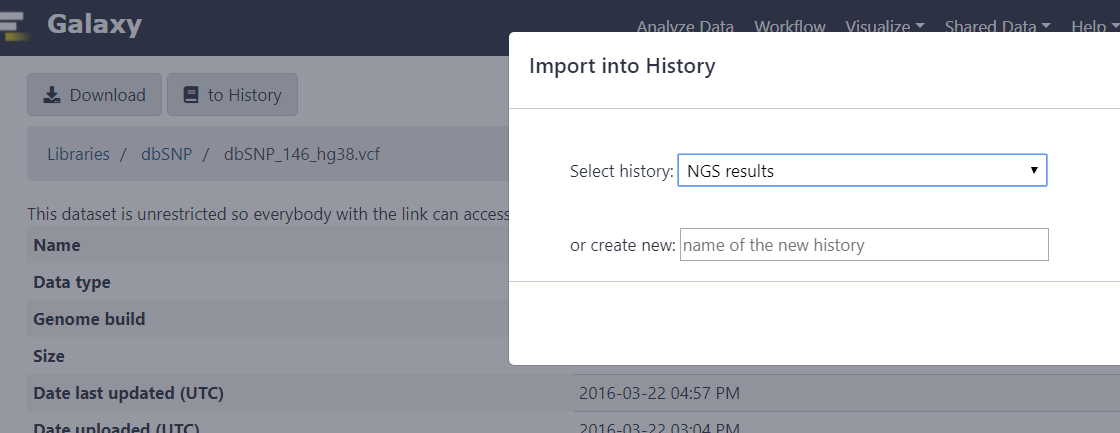
Сначала подключим нужную базу данных, перейдя по ссылкам Shared Data > Data Libraries.

В библиотеке выберем dbSNP, затем кликнем на версию dbSNP_146_hg38.vcf.
Кликнем на вкладке to History и выберем нашу историю.
Вернемся на страницу проекта и введем в строке поиска «dbsnp», а затем выберем в меню «SnpSift Annotate SNPs from dbSnp».
Здесь в первой строке выберем файл, который начинается с префикса SnpSift Variant Type, а во второй – загруженную базу данных. Выполняем действие.
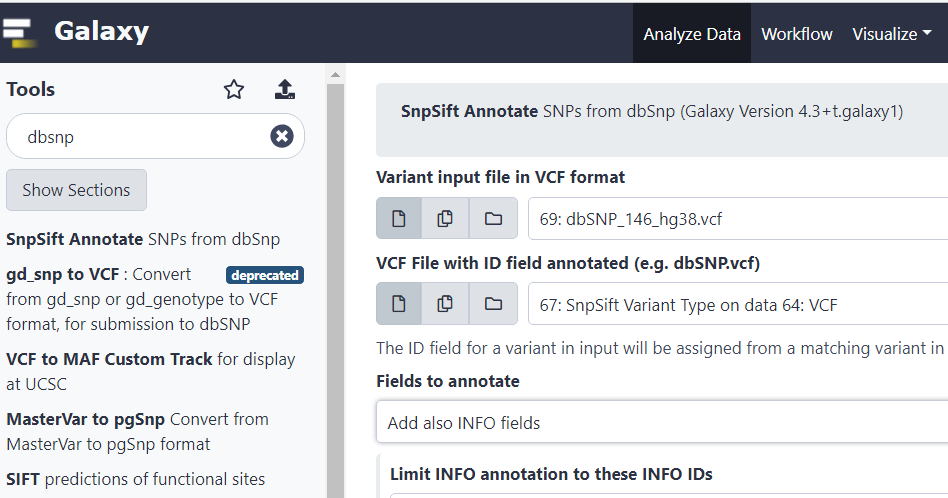
Теперь в нашем VCF-файле появились rs-записи в поле ID. Это уникальные RefSNP идентификаторы известных вариантов, о которых мы говорили выше. Также добавилось множество другой полезной информации в поле INFO.
Скачаем полученный VCF-файл (см. рисунок).
Что мы будем делать с VCF-файлом?
Конечно, Galaxy – прекрасное творение энтузиастов биоинформатики. Ее возможности огромны. Но, к сожалению, мне не удалось добиться совместимости с последними версиями баз данных Clinvar, dbNSFP, GWAS. А работать желательно с обновленными источниками. Поэтому для аннотирования VCF-файла мы спустимся с облачного сервиса на локальный компьютер.
Следующий вопрос, что же мы будем делать с VCF-файлом?
Первый этап – аннотирование VCF файла. В процессе аннотирования программа snpEff, точнее ее инструменты, автоматически добавят важную информацию. Аннотации включают описание ассоциированных заболеваний, прогноз влияния варианта на функцию гена, частоты аллелей и множество других полезных данных. Поскольку медицинские базы данных периодически дополняются, нам желательно использовать последние версии.
Второй этап – фильтрация и форматирование. На основе сделанных аннотаций, будут удалены данные, которые не влияют на функцию гена и скорее всего не имеют значения для здоровья. Результаты нужно будет сохранить в табличном виде.
Третья часть – сортировка и анализ табличных данных в Excel пои помощи инструментов интерпретации. Описанные нарушения нужно сравнить с проявлениями (фентоипом). Это самая трудоемкая, потому что «ручная» работа. Возможно, придется проверить сотни записей.
В следующих главах опробуем пройти весь этот длинный путь по порядку.
Можно сказать, что большая часть данных секвенирования избыточна, поскольку прочитанные варианты совпадают с эталонным геномом и неинтересны. Для анализа важны только различия, которые и записаны в VCF-файле.

Пропустив большую шапку, посмотрим на первые два столбика файла.
Под заголовком #CHROM идет порядковый номер одной из 23 пар хромосом, в которые упакованы нити ДНК.
Напомню, что 22 хромосомы парные (аутосомные), то есть одинаковы у мужчин и женщин. У мужчин кроме них есть непарные половые Х и Y-хромосомы. А у женщин есть две Х хромосомы, то есть, все 23 хромосомы парные. Хромосомы обозначают по порядковым номерам. Например, chr5 (5-я хромосома) или chrY (Y-хромосома).
POS – позиция прочитанной молекулы ДНК, одной из миллионов, по всей ее длине. Номера идут по возрастающей.
ID – обычно заполняется в процессе аннотирования уникальными номерами вариантов из «энциелопедии» dbSNP.
Разобравшись в предыдущей главе с аллелями, мы можем взглянуть на пятый и шестой столбики VCF файла, которые называются REF и ALT. Здесь REF – это заранее известный эталонный аллель, а ALT – найденные отличия от эталона, то есть, альтернативный аллель. Если альтернативными оказались оба гетерозиготных аллеля, то записывается их пара, разделенная запятыми.
Гетерозиготные варианты проявляются как позиции, где приблизительно половина чтений соответствует эталону, а другие показания отличаются от эталона.
0/1 – образец является гетерозиготным и содержит 1 копию каждого из аллелей – эталонного REF и альтернативного ALT
1/1 – образец является гомозиготным и отличается от эталона.
Сочетание 0/0 (гомозиготный эталонный) обычно не заносится в файл вместе с самой записью, потому что не имеет значения для анализа.
QUAL – качество. Это важный интегрированный параметр, который позволяет исключить варианты аллеля ALT, которые кажутся сомнительными. Как рассчитывается качество, я писал выше.
Аннотирование VCF-файла c программой snpEff
Аннотирование VCF-файла – ресурсоемкий процесс. Важно, чтобы на компьютере было установлено не менее 4Gb оперативной памяти (лучше 8Gb и больше). Мощный процессор, SSD диск и высокоскоростной интернет также существенно ускорят работу.
Работать придется не в Windows, а в Linux, потому что именно эту среду выбрали биоинформатики для своих проектов. Но не пугайтесь, если вы не сталкивались с Linux раньше. Мы установим не полноценный Linux, а эмулятор Cygwin, который прост в работе и должен нормально справиться с нашими задачами.
На эмуляторе мы установим snpEff. Это популярная и, на мой взгляд, лучшая программа для анализа генетических данных в свободном доступе. Хотя можно было бы работать и с другими программами, например, VEP или Annovar (но тут требуется регистрация на сервере некоммерческой организации в зоне .org).
Пользователям Linux я рекомендую параллельно смотреть эту ссылку. А для пользователей Windows предлагаю пошаговую видеоинструкцию.
Прежде всего, скачиваем и устанавливаем Cygwin в корень диска С (это важно!). Можно руководствоваться этим описанием установки.
После запуска Cygwin попросит установить логин и пароль. По имени логина будет создана папка, где будут хранится все файлы.
Скачаем последнюю версию программы Java и установим ее.
Заходим в Program Files, находим папку Java и копируем имя папки с версией программы.
Вставляем номер версии в строке вместо “jre1.8.0_261” (стараемся вставлять без пробелов, которые любит добавлять Word) или просто копируем мою строку, если имя вашей версии Java не отличается.
export PATH=$PATH:"/cygdrive/C/Program Files/Java/jre1.8.0_261/bin"Вставим эту строку в окно терминала Linux. Сразу обращу внимание, что в Cygwin вставка «ctrl+v» по умолчанию не работает, но можно пользоваться правым кликом и меню «вставить». Нажимаем Enter для ввода.
Далее проверяем, что Java запускается, для чего вводим тестовую команду на вывод версии:
java -versionЕсли в результате увидим java version с номером вашей версии, значит путь задан правильно и Java работает.

Скачиваем последнюю версию программы snpEff здесь.
Заходим в каталог установленной на диске С программы cygwin64, находим там папку home, а в ней папку с именем пользователя, которая была создана автоматически в процессе установки (в моем случае C:/cygwin64/home/Eugene). Распаковываем в нее архив snpEff_latest_core.zip. Появятся папки snpEff, clinEff и некоторые файлы, как на рисунке.

Пробуем запустить программу snpEff, вводим в командной строке
java -jar snpEff/snpEff.jarЕсли в результате мы увидим длинный текст, который начинается с ”SnpEff version SnpEff…”, то программа работает.
Создадим в той же папке, где находятся программы, каталог для VCF-файлов. Скопируем туда ваш VCF-файл и переименуем его в 01.vcf.
Теперь мы добавим первые 19 аннотаций (о них подробнее на английском тут).
Но прежде, чем вводить следующую команду, разберем и исправим ее при необходимости.
1) Важно правильно выбрать объем оперативной памяти, выделяемой компьютером на выполнение команд. Например, если у вашего компьютера лишь 4Gb оперативной памяти, то вместо параметра “-Xmx8g”, здесь и везде далее пишите “-Xmx4g”.
2) Сегодня параллельно используются две основные версии данных, точнее референсных генома, которые нужно четко разделять. В версии GRCh38/hg38, которая вышла в 2013 году, порядковый номер аллеля на хромосоме уже не советует предыдущей версии GRCh37/hg19. Это означает, что обязательно нужно выбирать соответствующие версии баз данных.
Чтобы выяснить, какая у вас версия, достаточно заглянуть в шапку VCF-файла. Если вы увидите в строках со словами reference или assembly «hg19», то референсный геном был GRCh37/hg19. Если же вы увидите «hg38», то референсный геном был GRCh38/hg38.
3) Важно правильно создавать структуру каталогов и прописывать пути к ним. Если вы допустите ошибку, вставите лишний пробел или длинное тире вместо знака минус, то программа выдаст ошибку.
Все, что написано ниже, относится к версии референсного генома GRCh38/hg38. Но отдельно в конце главы я продублировал команды и ссылки для версии GRCh37/hg19.
Также все, что написано ниже, относится к экзому. Вероятно, команды будут работать и с геномом, но будут пропущены некодирующие области ДНК.
Перейдем к первой команде аннотирования файла.
java -Xmx8g -jar snpEff/snpEff.jar -v -canon GRCh38.86 vcf/01.vcf> vcf/02.anncanon.vcf В папке vcf сразу появится выходной файл 02.anncanon.vcf. Но пока программа snpEff не загрузит базу данных размером более 600 Mb, его размер будет нулевым и с ним ничего происходить не будет. Сначала база будет загружаться в папку tmp программы Сygwin32. После загрузки она автоматически переместится в раздел data программы snpEff. В случае повторного обращения, база данных заново загружаться не будет, а будет сразу браться из папки data.
Интересно, что в процессе аннотирования наиболее сильно используются не ресурсы процессора, а память.

Чтобы убедиться, что файл был аннотирован, откроем в Notepad++ и сравним два варианта, пропустив всю шапку.
Добавились имя гена (OR4F5), тип варианта (synonymous_variant), степень влияния на функцию гена (LOW) и множество других аннотаций.
Чтобы сделать удобным будущий анализ в Excel, мы вывели выходной файл в каноническом структурированном виде (в строке за это отвечает установленный параметр “-canon”).
Нас очень интересует клиническая информация о наших вариантах. Чтобы ее получить, загрузим свежую версию базы данных Clinvar. Для референсного генома GRCh38/hg38 ее можно найти на сервере NCBI (известном как Pubmed), по этой ссылке.
Замечу, что иногда сервер блокирует IP определенных провайдеров, поэтому, если ссылка покажется не рабочей, включите какой-нибудь VPN (например, плагин Hotspot Shield Free VPN Proxy для Chrome).
Нас интересуют 3 файла (clinvar.vcf.gz, clinvar.vcf.gz.md5, clinvar.vcf.gz.tbi), которые нужно скачать. В каталоге snpEff сохраним эти файлы по пути: data/GRCh38/clinvar/ , для чего создадим соответствующие папки.

Теперь, если мы все сделали правильно, то можем аннотировать наш файл с Clinvar. Вводим:
java -Xmx8g -jar snpEff/SnpSift.jar annotate -v -info snpEff/data/GRCh38/clinvar/clinvar.vcf.gz vcf/02.anncanon.vcf > vcf/03.anncanon.clinvar.vcfБыли проаннотированы варианты, о которых известна какая-нибудь клиническая информация (в моем случае, около 7% записей VCF-файла).
Добавилось связанное с вариантом потенциальное заболевание, частота аллеля в популяции из проекта 1000 Genomes, характер влияния на потенциальное заболевание и многая другая информация.
Более подробно об аннотациях можно почитать здесь.
Вероятно, вам покажутся полезными аннотации из каталога GWAS (полногеномного поиска ассоциаций). Этот каталог не поможет найти редкие менделевские заболевания, но позволит выявить генетические факторы риска и дать прогноз о предрасположенности к распространенным заболеваниям и состояниям.
Замечу, что ссылка в инструкции к snpEff не работает, но каталог (размером 101Mb) можно скачать здесь.

Переименовываем файл “gwas_catalog_v1.0-associations_e100_r2020-06-30.tsv” в “gwascatalog.txt”, затем сохраняем его по такому пути: /db/GRCh37/gwasCatalog/gwascatalog.txt (потому что именно там его будет искать snpEff, несмотря на более свежую версию референсного генома).

Выполняем еоманду в терминале Linux.
java -Xmx8g -jar snpEff/SnpSift.jar gwasCat -v vcf/03.anncanon.clinvar.vcf > vcf/04.anncanon.clinvar.gwas.vcfВ моем случае было аннотировано 2.6% строк.
Следующая аннотация будет из базы данных dbNSFP. Она разработана для функционального прогнозирования вариантов в экзоме человека. Для большинства вариантов в кодирующей области, dbNSFP содержит десятки оценок, как на основе популяционных и филогенетических исследований (частота аллелей, консервативность участка), так и на основе моделей повреждения гена (например, MutationTester).
В инструкции к snpEff для референскного генома GRCh38 / hg38 рекомендуют использовать версию dbNSFP 3.2 Academic размером 14.4 Gb. На момент подготовки статьи ее можно было скачать по этой ссылке, а индексный файл, по этой ссылке.
В каталоге snpEff/data создадим папку dbnsfp и сохраним там оба файла. Затем выполним команду.
java -Xmx8g -jar snpEff/SnpSift.jar dbnsfp -v -db snpEff/data/dbnsfp/dbNSFP3.2a.txt.gz vcf/04.anncanon.clinvar.gwas.vcf > vcf/05.anncanon.clinvar.gwas.dbnsfp.vcfВ моем случае было аннотировано 7.75% данных.
Теперь мы удалим варианты низкой степени воздействия на функцию гена (LOW и MODIFIER), если только они не были аннотированы интересующими нас записями из баз данных Clinvar, GWAS, dbNSFP или dbSNP.
Все это мы сделаем при помощи команды filter:
java -Xmx8g -jar snpEff/SnpSift.jar filter -v " ( (ANN[0].IMPACT has 'HIGH') | (ANN[0].IMPACT has 'MODERATE') | (exists CLNSIGINCL) | (exists CLNDN) | (exists PMC) | (exists OM) | (exists MTP) | (exists TPA) | (exists MUT) | (exists GWASCAT_TRAIT) | (exists dbNSFP_MetaSVM_pred) | (exists dbNSFP_phastCons100way_vertebrate) | (exists dbNSFP_ExAC_NFE_AF) | (exists dbNSFP_Interpro_domain) ) " vcf/05.anncanon.clinvar.gwas.dbnsfp.vcf > vcf/06.anncanon.clinvar.gwas.dbnsfp.filtered.vcfСформируем таблицу, в которой оставим максимум информации и которую можно будет открыть в Excel.
java -Xmx8g -jar snpEff/SnpSift.jar extractFields -s "," -v vcf/06.anncanon.clinvar.gwas.dbnsfp.filtered.vcf CHROM POS ID REF ALT QUAL DP VARTYPE SNP MNP INS DEL MIXED HOM HET ANN[*].EFFECT ANN[*].IMPACT ANN[0].GENE ANN[1].GENE ANN[2].GENE LOF[*].PERC CLNSIG CLNDN CLNDISDB GWASCAT_TRAIT dbNSFP_MetaSVM_pred dbNSFP_Polyphen2_HDIV_pred dbNSFP_MutationTaster_pred dbNSFP_MutationAssessor_pred dbNSFP_Polyphen2_HVAR_pred dbNSFP_SIFT_pred dbNSFP_LRT_pred dbNSFP_PROVEAN_pred OM PMC MUT ANN[*].ALLELE ANN[*].GENEID ANN[*].FEATURE ANN[*].FEATUREID ANN[*].BIOTYPE ANN[*].RANK ANN[*].HGVS_C ANN[*].HGVS_P ANN[*].CDNA_POS ANN[*].CDNA_LEN ANN[*].CDS_POS ANN[*].CDS_LEN ANN[*].AA_POS ANN[*].AA_LEN ANN[*].DISTANCE ANN[*].ERRORS LOF[*].NUMTR NMD[*].NUMTR NMD[*].PERC DBVARID ALLELEID CDA OTH S3D WTD dbSNPBuildID SLO NSF R3 R5 NSN NSM G5A COMMON RS RV TPA CFL GNO VLD ASP ASS REF U3 U5 WGT MTP LSD NOC DSS SYN KGPhase3 CAF VC KGPhase1 NOV VP SAO INT G5 SSR RSPOS HD PM CLNVCSO CLNREVSTAT RS CLNDNINCL ORIGIN MC CLNVC CLNVI CLNSIGINCL GENEINFO CLNDISDBINCL CLNSIGCONF CLNHGVS SSR GWASCAT_P_VALUE GWASCAT_OR_BETA GWASCAT_REPORTED_GENE GWASCAT_PUBMED_ID CAF[1] CAF[2] AF_TGP[0] AF_EXAC[0] AF_ESP[0] dbNSFP_ExAC_NFE_AF[0] dbNSFP_ExAC_SAS_AF[0] dbNSFP_ExAC_Adj_AF[0] dbNSFP_1000Gp3_AMR_AF[0] dbNSFP_1000Gp3_EAS_AF[0] dbNSFP_ExAC_AFR_AF[0] dbNSFP_ExAC_AF[0] dbNSFP_ExAC_FIN_AF[0] dbNSFP_1000Gp3_EUR_AF[0] dbNSFP_ExAC_AMR_AF[0] dbNSFP_1000Gp3_AFR_AF[0] dbNSFP_ESP6500_AA_AF[0] dbNSFP_1000Gp3_SAS_AF[0] dbNSFP_ExAC_EAS_AF[0] dbNSFP_ESP6500_EA_AF[0] dbNSFP_1000Gp3_AF[0] dbNSFP_GERP___RS dbNSFP_GERP___NR dbNSFP_ExAC_Adj_AC dbNSFP_ExAC_SAS_AC dbNSFP_1000Gp3_AMR_AC dbNSFP_1000Gp3_EAS_AC dbNSFP_Interpro_domain dbNSFP_FATHMM_pred dbNSFP_ExAC_AFR_AC dbNSFP_1000Gp3_AC dbNSFP_ExAC_AC dbNSFP_ExAC_FIN_AC dbNSFP_phastCons100way_vertebrate dbNSFP_CADD_phred dbNSFP_1000Gp3_EUR_AC dbNSFP_ESP6500_EA_AC dbNSFP_1000Gp3_AFR_AC dbNSFP_ExAC_AMR_AC dbNSFP_ExAC_NFE_AC dbNSFP_1000Gp3_SAS_AC dbNSFP_ExAC_EAS_AC dbNSFP_ESP6500_AA_AC > vcf/extracted.txtСодержание:
Часть 1. Коротко о секвенировании
1.1. Когда делают секвенирование?
1.2. Что ожидать от результатов?
1.3. Что лучше экзом или геном?
Часть 2. Немного теории: чтение ДНК, аллели, поломки генов
2.1. Что и как секвенируют?
2.2. Аллели и наследственные менделевские заболевания
2.3. Что может поломаться в гене?
Часть 3. Обработка файлов секвенирования от А до Я
3.1. Выравнивание данных в Galaxy: от FASTQ к VCF-файлу
3.2. Что мы будем делать с файлами?
3.3. Аннотирование VCF-файла c программой snpEff
Часть 4. Инструменты интерпретации и анализ данных секвенирования в Excel
4.1. Инструменты интерпретации
4.1.1. Прогноз повреждения гена snpEff
4.1.2. Клиническая значимость от Clinvar
4.1.3. Частота аллеля (AF)
4.1.4. Консервативность участка
4.1.5. Аннотации dbSNP
4.2. Подготовка файла Excel
4.3. Анализ данных в Excel
